
Sponsored by
Allergan Global Medical Affairs

The information disclosed in this white paper is a result of a medical advisory board meeting held in Hawaii in January 2017. This white paper includes dry eye treatments in development by Allergan and is not inclusive of all therapies for DED that are currently under evaluation or in the early stages of research. This white paper is sponsored by Allergan Global Medical Affairs.

DED—DISRUPTION OF THE HOMEOSTATIC TEAR FILM
In 1995, the National Eye Institute, together with industry, organized a workshop with the intent to solidify a definition for dry eye disease (DED). The definition they generated stated, “Dry eye is a disorder of the tear film due to tear deficiency or excessive evaporation, which causes damage to the interpalpebral ocular surface and is associated with symptoms of ocular discomfort.” Since that time, understanding of the etiology, pathology, and treatment has increased considerably.
We now know that while multifactorial, DED is a chronic, immune-mediated inflammatory disease.1 When the delicate homeostatic balance of the ocular surface system is disturbed, it triggers the activation of a stress response that progresses from mitogen-activated protein kinases to transcription factors, and then production of proinflammatory cytokines and matrix metalloproteinases.2 This leads to the activation of T-cells that infiltrate the ocular surface and secrete additional proinflammatory cytokines.3,4 Thus begins a self-perpetuating cycle of inflammation and epithelial damage across the entire ocular surface, regardless of the origin of the disruption.
UNDERSTANDING THE TEAR FILM: T-CELLS ARE INTEGRAL TO TEAR FILM HOMEOSTASIS
It is important to understand the composition of the tear film and its interplay with the entire lacrimal functional unit. The innermost layer of the tear film is the mucin layer. The soluble mucin 5AC is produced by goblet cells in the conjunctiva and is essential for viscosity and stability during the blink cycle.5 Immediately external to the mucin is the aqueous layer produced by the lacrimal glands. While it is mostly a very diluted saltwater, it also contains a complex mixture of proteins, immunoglobulins, mucins, electrolytes, cytokines, lysozymes, lactoferrin, and growth factors, including T-cells.6 While we often speak of T-cells negatively, regulatory T-cells help inhibit a wide variety of autoimmune and inflammatory diseases,7 and their surface antigen, LFA-1, may play a critical role in regulatory T-cell homeostasis and function.8 The most abundant proteins, lysozyme and lactoferrin, possess antimicrobial functions. Immunoglobulins such as IgA, IgG, and IgM also have protective functions. Growth factors help regulate the processing of epithelial cell replacement and are necessary for wound healing. Electrolyte concentrations in healthy tears are carefully maintained within a certain range to ensure correct osmolarity, which is important for many aspects of epithelial and nerve cell function. The outermost lipid layer, produced primarily by the meibomian glands, restricts evaporation of the aqueous and lubricates the eye (Figures 1 and 2).
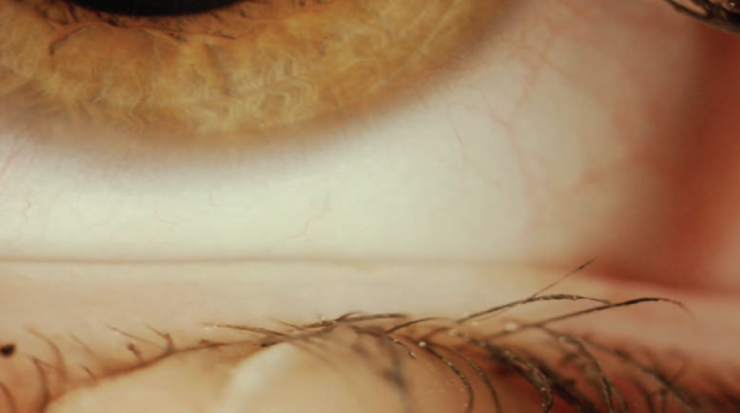
Figure 1. Patient with nonobvious MGD. Although the glands look healthy, they are plugged.
(Courtesy of P. Mile Brujic, OD, FAAO, and David L. Kading, OD, FAAO)
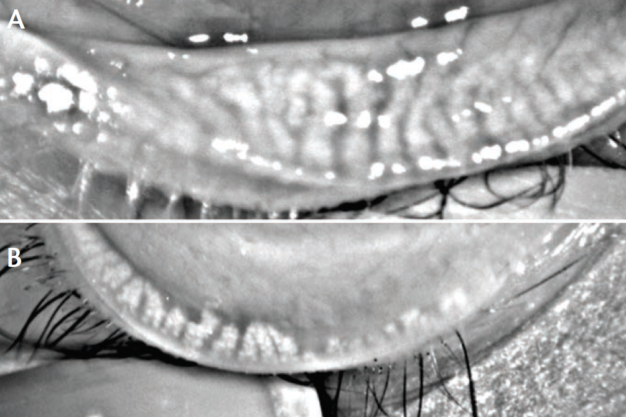
Figure 2. Eye with healthy meibomian glands (A); eye with meibomian gland dropout (B).
(Courtesy of P. Mile Brujic, OD, FAAO, and David L. Kading, OD, FAAO)
Tears in chronic DED are abnormal in many ways.5 A profound loss of goblet cells results in a less soluble and plentiful mucin 5AC, negatively impacting viscosity and adherence of the tear film. Tear protein concentrations, including those with antimicrobial functions, are reduced, along with growth factor concentrations. At the same time, proteases like matrix metalloprotease 9 (MMP-9) that are normally present in healthy tears in a constitutive low concentration in a latent, inactivated form become activated. They can degrade the extracellular matrix and the tight junctions between adjacent cells of the corneal epithelium. Activated proteases are also responsible for cleavage of many cytokines into an activated, proinflammatory form. Elevated tear electrolyte concentrations in DED parallel increased tear film osmolarity.
WHO HAS DED?
The triggers for DED are varied, but essentially fit into three categories. The first is physical irritation, such as environmental irritants, medications, contact lenses, lid abnormalities, or surgical trauma. The second category is systemic inflammation, including rheumatoid arthritis, lupus, or Sjogren syndrome. A third category of DED triggers is tear deficiency or instability arising from postmenopausal hormonal changes, or meibomian gland dysfunction.
Whatever the trigger, 26.4 million Americans reported suffering from DED9 in 2012, and many more are believed to suffer from DED yet do not report it or seek medical attention. The PHACO Study found that 87% of patients scheduled for cataract surgery had corneal staining, and 63% had a tear break-up time less than 5 seconds.10 This is alarming, as untreated DED can negatively impact preoperative measurements, healing, and comfort as well as visual outcomes.
Aside from the deleterious effect upon surgical outcomes, DED can have a significant negative impact on the quality of life of affected individuals. Severe DED is ranked similarly to severe angina and dialysis in validated patient surveys, and it impacts work performance, ability to drive at night, and enjoyment of outdoor activities.11,12 Furthermore, it is a common cause of contact lens intolerance.13
DIAGNOSING DED
DED pathogenesis is multifarious, presentation notoriously idiosyncratic, while signs and symptoms are commonly inconsistent, ruling out the possibility of a single diagnostic criteria. Most practitioners find it essential to deploy a variety of diagnostic tools to better define each patient’s disease state. Patient symptoms are generally documented by careful history, but can be measured according to functional visual quality with several standardized questionnaires such as the Ocular Surface Disease Index (OSDI) or the Symptom Assessment in Dry Eye (SANDE).
A proper examination must include careful evaluation of the ocular surface. Epithelial integrity is assessed with corneal and conjunctival staining, using fluorescein, rose Bengal or lissamine green (Figure 3). Impression cytology in a research setting is useful for measuring goblet cell density, inflammatory cell surface markers, and squamous metaplasia.
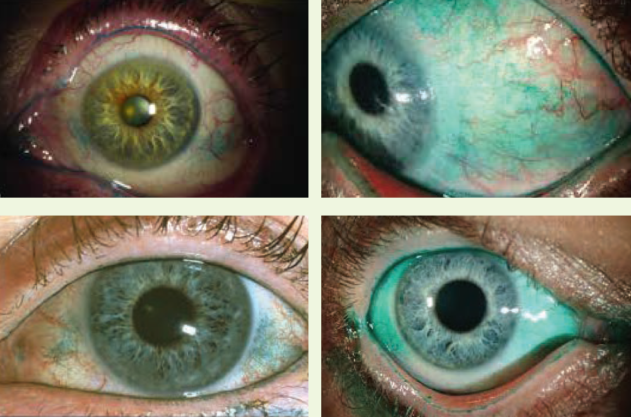
Figure 3. Lissamine green is used to identify DED and meibomian gland dysfunction.
(Courtesy of P. Dee G. Stephenson, MD)
Tear-based, point-of-service diagnostics currently available include tear secretion (Schirmer), tear osmolarity (TearLab), tear meniscus height (Oculus Keratograph 5M), tear film stability (tear break-up time), and MMP-9 protein elevation (InflammaDry, RPS). Meibomian gland function can be evaluated by lipid layer interferometry (Tear Science) and meibomian gland morphology readily documented by eyelid confocal microscopy or by DMI (dynamic meibomian imaging) (Tear Science, Oculus, Topcon). DMI is easily understood by patients who become far more engaged in their own treatment plan upon clear cut demonstration of their own anatomical changes. Corneal topography indices quantify the recognized effect of corneal irregularity upon visual quality and subsequent therapeutic response. The Sjogren syndrome test14 (Sjo, Bausch + Lomb, Immco Laboratories) detects early serum markers permitting much earlier identification of this syndrome that predisposes patients to lymphoma, pulmonary fibrosis, xerostomia, fatigue, and arthritis as well as severe DED.15
TREATING DED
Numerous extrinsic and intrinsic trigger factors for DED, combined with inconsistency in signs and symptoms, render effective therapy elusive. In 2003, the American Academy of Ophthalmology provided general guidelines for DED treatment that only briefly mention anti-inflammatory treatment.16 This treatment included cyclosporine and corticosteroids. By 2006, an international task force recommended specific treatments for each severity level that included a recommendation to consider topical cyclosporine for moderate to severe disease.17 In 2007, the International Dry Eye Workshop (DEWS) expanded on these guidelines and recommended topical cyclosporine for moderate or Level II disease.18 It was not until 2013 that the American Academy of Ophthalmology formally adopted the DEWS guidelines.19
It is now well understood that inflammation is one of the most important aspects of DED pathogenesis,20,21 and no matter the trigger, untreated or undertreated, established disease can lead to severe refractory disease.22 Atthis time, there are three topical prescription therapies available to treat inflammation in DED: corticosteroids, topical cyclosporine A (CsA), and lifitegrast. Oral essential fatty acid supplementation23 and tetracycline-class antibiotics24 are also commonly prescribed for inflammatory ocular conditions, including DED.
CORTICOSTEROIDS
While topical corticosteroids are not yet approved by the FDA specifically for the treatment of DED, they are potent anti-inflammatory drugs that are frequently prescribed off-label by eye care providers for induction25 and pulse therapy.26
Hydrocortisone is the main glucocorticoid secreted by the adrenal cortex, and its synthetic counterpart was first approved by the FDA in 1952 for the treatment of irritable bowel syndrome.27 The indication now includes more than 12 categories of use. The most widely used anti-inflammatory agents, corticosteroids suppress the molecular response to desiccating stress, which stimulates expression of MMP-9 and inflammatory cytokines while activating mitogen-activated protein kinase (MAPK) signaling pathways in the corneal epithelium.28 Essentially, steroids decrease inflammation by inhibiting T-cells on the ocular surface. Ocular and systemic corticosteroids also function by inducing phospholipase A2 inhibitory proteins that control the biosynthesis of inflammatory mediators, such as prostaglandins, ICAM, and leukotrienes, blunting diapedesis, and by inhibiting the release of their common precursor, arachidonic acid.
Difluprednate is a synthetic prednisolone derivate that was discovered in 1970. It was initially marketed in Japan by Senju Pharmaceuticals as a topical dermatologic formulation starting in 1979, and based on its binding affinity, it is considered a very strong steroid. Difluprednate 0.05% ophthalmic emulsion (Durezol; Alcon) was approved by the FDA for the treatment of inflammation and pain with ocular surgery in 2008 and anterior uveitis in 2009.29
CYCLOSPORINE A
Cyclosporine is a fungal-derived peptide that was first approved by the FDA in 1983 to suppress the immune system in all organ transplant patients (Sandimmune, Sandoz Pharmaceuticals).30 The FDA approved a microemulsion formulation of cyclosporine (Neoral, Novartis) in 1995 and Gengraf (AbbVie) in 2000. Cyclosporine A was first used topically in ophthalmology in the early 1980s to inhibit experimental corneal allograft rejection, and was later found useful for various ocular inflammatory disorders. It was first tested in conjunction with keratoconjunctivitis sicca in canines in 1989,31 prior to studies by Leibovitz showing efficacy in humans.32 Restasis (cyclosporine A 0.05% ophthalmic emulsion; Allergan) was approved by the FDA in 2002 to increase tear production in patients whose tear production is presumed to be suppressed due to ocular inflammation associated with dry eye.
Cyclosporine was the first immunosuppressive drug that allowed for selective regulation of T-cells while preserving ocular resident epithelial cells from inflammatory apoptosis through differential modulation of MPTP activity.33 Cyclosporine also attenuates responses of immune as well as nonimmune cells to inflammatory stimuli via suppression of nuclear factor kB (NF-kB) transcriptional signaling. It has been well documented that cyclosporine selectively inhibits T-cell activation through blocking Ca2þ-activated, calcineurin-dependent nuclear factor of activated T-cells (NFAT), and subsequently blocks cytokine (such as IL-2) and chemokine production.34 NF-kB upregulates the production of inflammatory mediators, and is one of the most important factors in proinflammatory gene expression,35 as well as plays a key role in apoptosis, stress response, corneal wound healing, angiogenesis, and lymph-angiogenesis.36
Creation of an appropriate vehicle for ophthalmic delivery of insoluble and hydrophobic cyclosporine was necessary, so accordingly Allergan developed an emulsion formulation in castor oil that also includes glycerin, polysorbate 80, and sodium hydroxide.37 The phase 3 studies of this topical emulsion (Restasis) showed a demonstrated treatment effect after the first month that increased over 6 months with improvements in corneal staining, Schirmer values, blurred vision, and physician evaluation.38
Importantly, topical administration of cyclosporine A 0.05% or 0.1% ophthalmic emulsions resulted in undetectable or very low plasma levels of cyclosporine. Following topical administration of cyclosporine A 0.05% ophthalmic emulsion twice daily for up to 12 months, blood concentrations of cyclosporine remained below the quantitation limit of 0.1 ng/mL. Serum concentrations detected were several orders of magnitude less than those found during systemic immunosuppressive applications.39
LFA-1 INHIBITORS
Leukocytes migrate to sites of inflammation by engaging their ligands on endothelial cells, and many immune-mediated chronic inflammatory diseases, including rheumatoid arthritis, psoriasis, and inflammatory bowel disease, are characterized by this unregulated migration.40 Targeting integrins and ligands of the immunoglobulin superfamily, which are responsible for leukocyte migration, is thought to modulate inflammation and has successfully been used to treat inflammatory bowel diseases.41
The integrin lymphocyte function-associated antigen 1 (LFA-1) and its interaction with the ligand ICAM-1 is necessary for the trafficking of inflammatory cells.42 A humanized monoclonal antibody that targeted the integrin L, was previously on the market (efalizumab, Raptiva) to treat plaque psoriasis. It prevented LFA-1 expressing lymphocytes from interacting with ICAM-1 ligands, thus preventing activation of T-cells and their migration to the skin. Efalizumab was approved in 2003 for treatment of plaque psoriasis and a subsequent safety study showed that patients could receive 1 mg/kg subcutaneously for 24 weeks with no evidence of cumulative toxic effects.43 However, in April 2009 efalizumab was voluntarily withdrawn from the US market due to the report of three cases of progressive multifocal leukoencephalopathy (PML) in patients with psoriasis. PML is a rare central neurological disease caused by the JC polyomavirus (JCV) that has been associated with several monoclonal antibodies intended to treat chronic inflammatory diseases, including Rituxan (Genentech) and other biologics.44 JCV is carried by approximately 58% of the population,45 but it is kept in check by T-cells, which are the most important component of the immune response against established intracellular viruses.46 Untreated PML has a 50% mortality rate47 and occurs when the cellular immunity is repressed, thereby markedly decreasing CD4+ T-cell counts.
In 2010, a novel series of small molecules derived from tetrahydroisoquinoline (THIQ) were discovered that could disrupt binding of LFA-1 to its receptor ICAM-1, which demonstrated good bioavailability with either oral or IV administration.48 Lifitegrast was identified from this series of THIQ-derived small molecules and engineered as a room temperature stable, pH neutral, highly soluble topical ophthalmic solution.49,50 Lifitegrast inhibits the interaction of LFA-1 and ICAM-1, and thus the subsequent cycle of T-cell–mediated inflammation. This includes inhibiting neutrophil recruitment to the corneal stroma51 and inhibiting cytokine release from activated lymphocytes.52
The safety and efficacy of lifitegrast 5% solution (Xiidra, Shire Pharmaceuticals) has been assessed in a total of 1,181 patients in four, 12-week, randomized, multicenter, double-masked, vehicle-controlled studies.53 The studies found that individuals treated with Xiidra demonstrated greater improvement in signs and symptoms of DED than in groups treated with phosphate buffered saline.54,55 On July 12, 2016, Xiidra was approved for treatment of the signs and symptoms of DED under the lymphocyte function-associated antigen 1 antagonist category. The most common side effects of lifitegrast 5% solution include eye irritation, blurred vision, and dysgeusia. In a subset of DED patients enrolled in phase 3 trials for lifitegrast 5% solution, 19% (9/47) had plasma lifitegrast trough concentrations above the lower limit of assay quantitation (0.5 ng/mL), ranging from 0.55 ng/mL to 3.74 ng/mL.56
NEUROSTIMULATION AND DED
While lifitegrast 5% solution broke ground with a class of drug new to DED treatment, the dry eye market continues to expand and gaps in care remain. The next device to come to market will be an alternative to pharmacotherapy or surgery: neurostimulation.
Neurostimulation is an established, yet highly innovative field that provides an alternative to pharmacotherapy or surgical procedures in a wide variety of conditions. This unique approach to therapy involves applying stimulation techniques to the affected region of the nervous system to alter neurophysiological signals affecting target tissues and organs.57 The first implantable neurostimulator for relief of chronic pain was approved in 1967,58 and subsequent devices have treated millions of patients for myriad conditions including tremors,59 epilepsy,60 obsessive-compulsive disorder,61 Parkinson disease,62 obesity,63 overactive bladder, 64 and retinitis pigmentosa.65 Neurostimulators today are either surgically implanted, usually near a central or peripheral nerve, or employ portable technologies that conduct the stimulation transcutaneously or percutaneously.66
The lacrimal functional unit (LFU) consists of the lacrimal glands, ocular surface, and lids, regulated by sensory and motor innervation. This is an integrated system whose interdependent components act together, and not in isolation.67 Abnormality in any of the extensive neural connections can result in an altered tear film, which no longer supports the normal functioning of the ocular surface. Damage to lacrimal gland innervation can result from diseases such as Sjogren syndrome, refractive surgery, chronic use of contact lens, ocular herpes simplex or zoster infection, adenovirus keratoconjunctivitis, or simply aging; the result is decreased tear secretion.68
Upon stimulation, the LFU communicates with the afferent trigeminal nerve via sensory neurons carrying information from the ocular surface, glands, and tissues to the central nervous system, while efferent parasympathetic and sympathetic neurons carry information from the central nervous system to the LFU.69 The trigeminal nerve is the largest cranial nerve and has three branches, the ophthalmic nerve, the maxillary nerve, and the mandibular nerve. The ophthalmic nerve is responsible for innervation of the LFU, including the lacrimal gland, the meibomian glands, and the goblet cells.70,71
An intranasal tear neurostimulator, TrueTear (Allergan) is currently awaiting FDA review and approval for treatment of DED. The noninvasive device has two disposable prongs that are inserted into the nose to contact nasal mucosal fibers of the nasociliary branch of the trigeminal nerve. A highly refined low level electrical pulse stimulates the afferent cranial nerve Vii, which then communicates through the brainstem to efferent trigeminal fibers controlling the lacrimal gland, ocular surface goblet cells, and tarsal meibomian glands, thereby increasing truly natural, not reflex, tear production.
CLINICAL APPROACH TO THE DRY EYE PATIENT
Eye care providers who recognize and treat dry eye generally encounter three typical scenarios. Those who do not should at least refer to an interested colleague within their practice or community.
(1) A new patient, often referred, presenting for the treatment of dry eye, usually having seen one or more previous clinicians and failed several different treatments. This patient requires a careful diagnostic evaluation with step wise, logical, scientific introduction of one therapy at a time. Adequate time must be allowed for full treatment effect: at least 6 weeks. It is wise to introduce one rapid onset therapy, such as topical steroids or punctal plugs, plus one slower onset therapy, such as nutritionals, lid scrubs, or thermal pulsation, thereby allowing both patient and clinician to differentiate between the two interventions.
(2) An existing patient with another primary diagnosis who is identified with dry eye. Careful introduction of new treatments is again warranted, with careful attention to minimizing excessive drop use, particularly in glaucoma patients. These clinic visits may require more than the usual visit time due to their complexity, so the temptation to shrug off significant punctate keratopathy or increased discomfort until the next visit should be suppressed. Some elderly or neurotrophic patients with obvious ocular surface damage experience minimal discomfort and require more thorough explanations and intensive encouragement.
(3) A new or current patient preparing for ophthalmic surgery. Meticulous attention must always be paid to the condition of the refractive surface in order to optimize biometry, healing, and surgical outcomes. In this situation, accelerated ocular surface normalization is preferred, and multiple complementary therapies may be simultaneously initiated. Many cataract, corneal refractive, glaucoma, retina, oculoplastic, and corneal transplant patients may receive expeditious recommendations for all or most of the following in order to hasten response time: oral nutritionals, preservative-free tears, lid scrubs containing hypochlorous acid (Avenova, Hypochlor), punctal plugs, oral doxycycline, thermal pulsation, and topical loteprednol, cyclosporine, or lifitegrast.
Patient-centered recommendations reflect the urgency, etiology, clinical context, and severity of each patient presentation. Treatment paradigms are directed by each unique set of signs and symptoms, history and review of systems, point-of-service testing, and then intelligent treatment prioritization. It is only through complete efforts to collect all relevant data that the best possible treatment can be selected.
CONCLUSION
DED affects millions of Americans,72 and the symptoms range from occasional mild discomfort to debilitating.73 Science has come a long way in understanding the underlying etiology of this disease, creating novel interventions and adapting systemic therapeutics to topical formulations specifically targeting the underlying DED process.
1. Stevenson W, Chauhan SK, Dana R. Dry eye disease: an immune-mediated ocular surface disorder. Arch Ophthalmol. 2012;130(1):90-100.
2. Li DQ, Luo L, Chen Z, Kim HS, Song XJ, Pflugfelder SC. JNK and ERK MAP kinases mediate induction of IL-1, TNF-a and IL-8 following hyperosmolar stress in human limbal epithelial cells. Exp Eye Res. 2006;82(4):588-596.
3. De Paiva CS, Chotikavanich S, Pangelinan SB, et al. IL-17 disrupts corneal barrier following desiccating stress. Mucosal Immunol. 2009;2(3):243-253.
4. El Annan J, Chauhan SK, Ecoiffier T, Zhang Q, Saban DR, Dana R. Characterization of effector T cells in dry eye disease. Invest Ophthalmol Vis Sci. 2009;50(8):3802-3807.
5. Pflugfelder SC, Beuerman RW, Stern ME, eds. Dry Eye and Ocular Surface Disorders. New York, NY: Marcel Dekker; 2004.
6. Prabha JL. Tear secretion—a short review. J Pharm Sci Res. 2014;6(3):155-157.
7. Siemasko KF, Gao J, Calder VL, et al. In vitro expanded CD4+ CD25+ Foxp3+ regulatory T cells maintain a normal phenotype and suppress immune-mediated ocular surface inflammation. IOVS. 2008;49:5434-5440.
8. Wohler J, Bullard D, Schoeb T, Carnum S. LFA-1 is critical for regulatory T cell homeostasis and function. Mol Immunol. 2009;46(11-12):2424-2428.
9. The Gallup Organization, Inc. The 2012 Gallup Study of Dry Eye Sufferers. 2012.
10. Trattler WB, Reilly CD, Goldberg DF, et al. Cataract and dry eye: prospective health assessment of cataract patient ocular surface (PHACO) study. Paper presented at: ASCRS Symposium and Congress; May 25-29, 2011; San Diego, CA.
11. Buccholz P, Steeds CS, Stern LS, et al. Utility assessment to measure the impact of dry eye disease. Ocul Surf. 2006 Jul;4(3):155-161.
12. Yamada M, Mizuno Y, Shigeyasu C. Impact of dry eye on work productivity. Clinicoecon Outcomes Res. 2012;4: 307-312.
13. McMahon TT, Zadnik K. Twenty-five years of contact lenses: the impact on the cornea and ophthalmic practice. Cornea. 2000;19:730-740.
14. Shen L, Suresh L, Lindemann M, et al. Novel autoantibodies in Sjögren’s syndrome. Clin Immunol. 2012;145:251-255.
15. Akpek EK, Mathews P, Hahn S, et al. Ocular and systemic morbidity in a longitudinal cohort of Sjögren’s syndrome. Ophthalmology. 2015;122(1):56-61.
16. American Academy of Ophthalmology Cornea/External Disease Panel. Preferred Practice Pattern® for Dry Eye Syndrome. San Francisco, CA: American Academy of Ophthalmology; 2003.
17. Behrens A, Doyle JJ, Stern L, et al; Dysfunctional Tear Syndrome Study Group. Dysfunctional tear syndrome: a Delphi approach to treatment recommendations. Cornea. 2006;25(8):900-907.
18. Definition and Classification Subcommittee. The definition and classification of dry eye disease: report of the Definition and Classification Subcommittee of the International Dry Eye Workshop. Ocul Surf. 2007;5(2):75-92.
19. American Academy of Ophthalmology Cornea/External Disease Panel. Preferred Practice Pattern® for Dry Eye Syndrome. San Francisco, CA: American Academy of Ophthalmology; 2013.
20. Calonge M, Enríquez-de-Salamanca A, Diebold Y, et al. Dry eye disease as an inflammatory disorder. Ocul Immunol Inflamm. 2010;18:244-253.
21. Stevenson W, Chauhan SK, Dana R. Dry eye disease: an immune-mediated ocular surface disorder. Arch Ophthalmol. 2012;130: 90-100.
22. Baudouin C. The vicious circle in dry eye syndrome: a mechanistic approach. J Fr Ophtalmol. 2007;3:239-246.
23. Sheppard JD, Singh R, McClellan AJ, et al. Long-term supplementation with n-6 and n-3 PUFAs improves moderate- to-severe keratoconjunctivitis sicca: a randomized double-blind clinical trial. Cornea. 2013;32(10):1297-1304.
24. De Paiva CS, Corrales RM, Villarreal AL, et al. Corticosteroid and doxycycline suppress MMP-9 and inflammatory cytokine expression, MAPK activation in the corneal epithelium in experimental dry eye. Exp Eye Res. 2006;83:526-535.
25. Sheppard JD, Donnenfeld ED, Holland EJ, et al. Effect of loteprednol etabonate 0.5% on initiation of dry eye treatment with topical cyclosporine 0.05%. Eye Contact Lens. 2014;40(5):289-296.
26. Marsh P, Pflugfelder SC. Topical non-preserved methylprednisolone therapy of keratoconjunctivitis sicca in Sjogren’s syndrome. Ophthalmology. 1999;106:811-816.
27. FDA. https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=008697. Accessed April 4, 2017.
28. De Paiva CS, Corrales RM, Villarreal AL, et al. Corticosteroid and doxycycline suppress MMP-9 and inflammatory cytokine expression, MAPK activation in the corneal epithelium in experimental dry eye. Exp Eye Res. 2006;83(3):526- 535.
29. Sheppard JD, Toyos MM, Kempen JH, et al. Difluprednate 0.05% versus prednisolone acetate 1% for endogenous anterior uveitis: a phase III, multicenter, randomized study. Invest Ophthalmol Vis Sci. 20146;55(5):2993-3002.
30. AP. http://www.nytimes.com/1983/09/03/us/drug-that-reduces-risk-in-transplants-gets-early-approval.html. Published September 3, 1985. Accessed April 4, 2017.
31. Kaswan R, Salisbury M, Ward D. Spontaneous canine keratoconjunctivitis sicca. A useful model for human keratoconjunctivitis sicca: treatment with cyclosporine eye drops. Arch Ophthalmol. 1989;107:1210-1216.
32. Laibovitz RA, Solch S, Andrianao J. Pilot trial of cyclosporine 1% ophthalmic ointment in the treatment of keratoconjunctivitis sicca. Cornea. 1993;12:315-323.
33. Gao J, Sana R, Calder V, et al. Mitochondrial permeability transition pore in inflammatory apoptosis of human conjunctival epithelial cells and T cells: effect of cyclosporin A. Invest Ophthalmol Vis Sci . 2013; 54(7): 4717-4733.
34. Donnenfeld E, Pflugfelder SC. Topical ophthalmic cyclosporine: pharmacology and clinical uses. Surv Ophthalmol. 2009;54(3):321-338.
35. Tak, P; Firestein P. NF-kB: a key role in inflammatory diseases. The Journal of Clinical Investigation. 2001;107:7-11.
36. Lan W, Petznick A, Heryati S, et al. Nuclear factor-kB: Central regulator in ocular surface inflammation and diseases. Ocul Surf. 2012;10(3):137-48.
37. Restasis Package Insert. Allergan, Inc. 2004
38. Sall K, Stevenson OD, Mundorf TK, et al. Two multicenter, randomized studies of the efficacy and safety of cyclosporine ophthalmic emulsion in moderate to severe dry eye disease. Ophthalmology. 2000;107:631-639.
39. Tang-Liu DD, Acheampong A. Ocular pharmacokinetics and safety of cyclosporine, a novel topical treatment for dry eye. Clin Pharmacokinet. 2005;44:247-261.
40. Davidson A, Diamond B. Autoimmune diseases. N Engl J Med. 2001;345:340-350.
41. Bamias G, Clark DJ, Rivera-Nieves J. Leukocyte traffic blockade as a therapeutic strategy in inflammatory bowel disease. Curr Drug Targets. 2013;14(12):1490-1500.
42. Shang XZ, Issekutz AC. Contribution of CD11a/CD18, CD11b/CD18, ICAM-1 (CD54) and -2 (CD102) to human monocyte migration through endothelium and connective tissue fibroblast barriers. Eur J Immunol. 1998;28(6):1970- 1979.
43. Menter A, Gordon K, Carey W, et al. Efficacy and safety observed during 24 weeks of efalizumab therapy in patients with moderate to severe plaque psoriasis. Arch Dermatol. 2005;141(1):31-8.
44. Toussirot É, Bereau M. The risk of progressive multifocal leukoencephalopathy under biological agents used in the treatment of chronic inflammatory diseases. Inflamm Allergy Drug Targets. 2014;13(2):121-127. Review.
45. Egli A, Infanti L, et.al. Prevalence of polyomavirus BK and JC infection and replication in 400 healthy blood donors. J Infect Dis. 2009;199(6): 837-846.
46. Koralnik IJ. Overview of the cellular immunity against JC Virus in progressive multifocal leukoencephalopathy. J NeuroVirol. 2008;8(suppl.2):59-65.
47. Brew BJ, Davies NW, Cinque P, et al. Progressive multifocal leukoencephalopathy and other forms of JC virus disease. Nat Rev Neurol. 2010; 6(12):667-679.
48. Zhong M, Shen W, Barr KJ, et al. Discovery of tetrahydroisoquinoline (THIQ) derivatives as potent and oraly bioavailable LFA-1/ICAM-1 antagonists. Bioorg Med Chem Lett. 2010 1;20(17):5269-5273.
49. Perez VL, Pflugfelder SC, Zhang S, et al. Lifitegrast, a novel integrin antagonist for treatment of dry eye disease. Ocul Surf. 2016;14(2):207-215.
50. Semba CP, Gadek TR. Development of lifitegrast: a novel T-cell inhibitor for the treatment of dry eye disease. Clin Ophthalmol. 2016;10: 1083-1094.
51. Sun Y. Zhang R, Gadek TR, et al. Corneal inflammation is inhibited by the LFA-1 antagonist, lifitegrast (SAR 1118). J Ocul Pharmacol Ther. 2013;29: 395-402.
52. Murphy CJ, Bentley E, Miller PE, et al. The pharmacologic assessment of a novel lymphocyte function-associated antigen-1 antagonist (SAR 1118) for the treatment of keratoconjunctivitis sicca in dogs. Invest Ophthalmol Vis Sci. 2011; 52:3174-3180.
53. XIIDRA Package Insert. Lexington, MA: Shire US Inc.; 2016.
54. Sheppard J,Torkildsen GL, Lonsdale JD, et al. Lifitegrast ophthalmic solution 5.0% for treatment of dry eye disease: results of the OPUS-1 phase 3 study. Ophthalmology. 2014;121(2):475-483.
55. Tauber J, Karpecki P, Latkany R, et al. Lifitegrast ophthalmic solution 5.0% versus placebo for teatment of dry eye disease: Results of the randomized phase III OPUS-2 Study. Ophthalmology. 2015;122: 2423-2431.
56. Xiidra Prescribing Information. http://www.accessdata.fda.gov/drugsatfda_docs/label/2016/208073s000lbl.pdf Last viewed March 1, 2017.
57. Danilov YP, Kublanov VS. Emerging noninvasive neurostimulation technologies: CN-NINM and sympatocorection. J Behav Brain Sci. 2014;4:105-113.
58. Mekhail NA, Cheng J, Narouze S, et al. Clinical applications of neurostimulation: forty years later. Pain Pract. 2010;10(2):103-112.
59. Karas PJ, Mikell CB, Christian E, et al. Deep brain stimulation: a mechanistic and clinical update. Neurosurg Focus. 2013;35(5):e1.
60. Rielo DA. Vagus nerve stimulation. Medscape website. http://emedicine.medscape.com/article/1186123- overview. Updated November 17, 2015. Accessed September 4, 2016.
61. Reclaim DBS therapy for OCD-H050003. US Food and Drug Administration website.http://www.fda.gov/MedicalDevices /ProductsandMedicalProcedures/DeviceApprovalsandClearances/Recently ApprovedDevices/ucm125520. htm. Updated September 6, 2013. Accessed September 4, 2016.
62. Gardner J. A history of deep brain stimulation: Technological innovation and the role of clinical assessment tools. Soc Stud Sci. 2013;43(5):707-728.
63. EnteroMedics Maestro rechargeable system-P130019. US Food and Drug Administration website. http://www. fda.gov /MedicalDevices/ProductsandMedicalProcedures/DeviceApprovalsandClearances/RecentlyApprovedDevices/ ucm430696.htm. Updated January 16, 2015. Accessed September 4, 2016.
64. Staskin DR, Peters KM, MacDiamid S, et al. Percutaneous tibial nerve stimulation: a clinically cost effective addition to the overactive bladder algorithm of care. Curr Urol Rep. 2012:13(5):327-334.
65. Argus II retinal prosthesis system-H110002. US Food and Drug Administration website. http://www.fda. gov /MedicalDevices/ProductsandMedicalProcedures/DeviceApprovalsandClearances/Recently-ApprovedDevices/ ucm343162.htm. Updated January 13, 2015. Accessed September 4, 2016.
66. Henry R, Deckert M, Guruviah V, Schmidt B. Review of neuromodulation techniques and technological limitations. IETE Tech Rev. 2015; Nov. http://dx.doi.org/10.1080/02564602.2015.1106926. Accessed September 4, 2016.
67. Beuerman RW, Mircheff A, Pflugfelder SC, Stern ME. The lacrimal functional unit. In: Pflugfelder SC, Beuerman RW, Stern ME, eds. Dry Eye and Ocular Surface Disorders. 1st ed. Marcel Dekker, Inc.; 2004;11-39.
68. Dartt DA. Dysfunctional neural regulation of lacrimal gland secretion and its role in the pathogenesis of dry eye syndromes. Ocul Surf. 2004;2(2):76-91.
69. Kossler AL, Wang J, Feuer W, Tse DT. Neurostimulation of the lacrimal nerve for enhanced tear production. Ophthal Plast Reconstr Surg. 2015;31(2):145-151.
70. Waxman SG. Chapter 8. Cranial Nerves and Pathways. In: Waxman SG. eds. Clinical Neuroanatomy, 27e. New York, NY: McGraw-Hill; 2013. http://accessmedicine.mhmedical.com.ezproxy.agnlib.com/content.aspx?bookid=673&Sectionid=45395970.
71. Brinton M, Cheng JL, Kossler A, et al. Electronic enhancement of tear secretion. J Neural Eng. 2016;13(1):016006.
72. National Eye Institute. https://nei.nih.gov/health/dryeye/dryeye
73. Sheppard JD. Dry eye syndrome. Managed Care.2003;12(12):supplement.


