




Since its inception, the promise of microinvasive glaucoma surgery (MIGS) has been to address a large unmet need for patients who do not require the more potent, invasive, and risky traditional filtering surgery but are not best served by topical medications or laser trabeculoplasty. By addressing quality of life, adherence, and IOP lowering in a safe manner and with more rapid recovery, MIGS procedures have found an important niche, both combined with cataract surgery and as a standalone option. MIGS was put forward as a surgical category with a high degree of safety, so the bar was set high from the start.
The recent news of Alcon’s voluntary global market withdrawal of its CyPass Micro-Stent due to concerns regarding endothelial cell loss (ECL) has raised many unanswered questions. To date, there is no evidence to suggest that ECL is an issue with trabecular bypass and Schlemm canal–based devices. As the MIGS wheels of change continue to encourage early intervention in glaucoma, the newly released results of Alcon’s COMPASS-XT trial (available at www.alcon.com/cypass) have shifted attention back to this question: Are these surgeries safe enough?
A favorable risk profile is one of the main characteristics of MIGS devices. Surgeons are willing to obtain modest IOP lowering compared with traditional glaucoma surgeries if they feel confident that the risk of sight-threatening complications is minimal. Does this remain true with the recent news of ECL reported by Alcon for the CyPass Micro-Stent? This development raises certain questions: What went wrong, how can we remedy it, and does the same risk apply to other MIGS devices?
WHAT DO WE KNOW ABOUT THE CYPASS WITHDRAWAL?
In early September, Alcon announced the voluntary global market withdrawal of the CyPass Micro-Stent due to concerns of progressive ECL. Any time patient safety is of concern, erring on the side of caution is the preferred approach. We applaud Alcon’s swift handling of the situation.
The COMPASS-XT trial was a 3-year extension of Alcon’s 2-year COMPASS trial comparing cataract surgery with CyPass implantation (study eyes) and cataract surgery alone (control eyes) in patients with open-angle glaucoma. The COMPASS trial was the pivotal study on which FDA approval of the CyPass Micro-Stent was based. The COMPASS-XT trial included 282 of the 505 patients enrolled in the COMPASS trial, and 253 patients completed the 60-month visit. In the initial 24-month data, both the mean percentage of ECL and the percentage of eyes with significant ECL (>30%) were comparable between the two groups. The FDA required Alcon to continue following ECL after market approval.
Early analysis of the COMPASS-XT trial at 5 years (data provided by the company) showed significantly more central ECL in the cataract surgery plus CyPass group than the control group. Baseline endothelial cell density (ECD) was 2,432 cells/mm2 for CyPass patients and 2,434 cells/mm2 for control patients. At 60 months, ECD decreased to 1,931 cells/mm2 (-18.4%) in the CyPass group (n = 163) compared with 2,189 cells/mm2 (-7.5%) in the control group (n = 40; Figure 1). At 60 months, significant ECL (>30%) was more common in the CyPass group (27.2%) compared with the control group (10%; Figure 2).
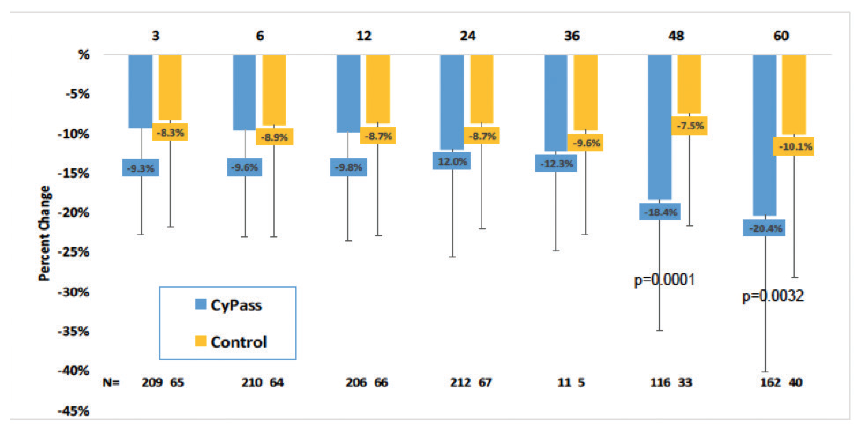
Figure 1. Percentage of change in ECD from baseline.
(Courtesy of Alcon)
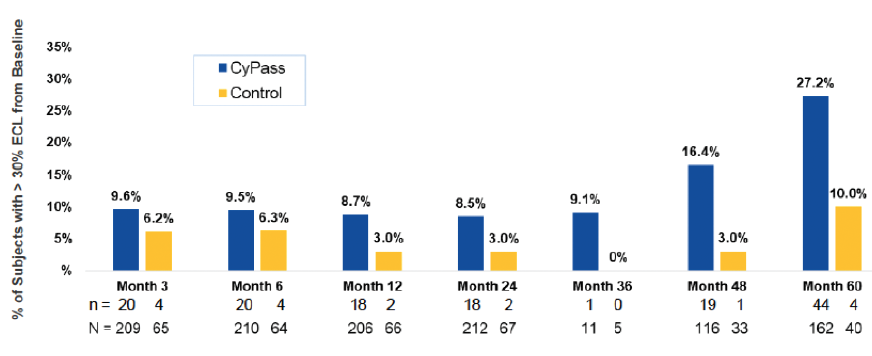
Figure 2. Percentage of patients with >30% ECL from baseline.
A number of variables were assessed to determine their correlation with ECL. The device position in the anterior chamber angle was the only factor in the analysis that correlated with ECL. The ideal position appears to be with no retention rings visible and with the top of the device flush with the trabecular meshwork. In the COMPASS-XT trial, yearly ECL rates were 1.39% when no ring was visible (similar to controls), compared with 2.74% when one ring was visible and 6.96% when two or three rings were visible (Figure 3). Interestingly, some patients with two or more visible rings did not have significant ECL, which makes us suspect that there are other factors in play, such as the angulation of the device in the anterior chamber, which can vary based on scleral curvature. Only one patient developed corneal edema, assessed as mild, at 51 months. Endothelial touch was detected, and the device was successfully trimmed 4 months later, with subsequent resolution of the edema at study completion.
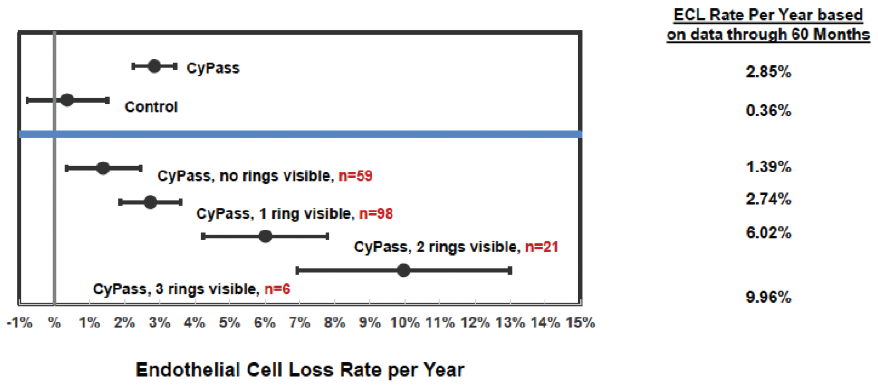
Figure 3. Annualized ECL at 60 months by device position.
Although it is possible that other variables—such as material, change in aqueous flow, and reflux flow—play a role in ECL with CyPass, there is no evidence of this yet. Further, given the strong correlation with mechanical position of the implant in the anterior chamber (and that more deeply implanted devices had similar ECL to controls), this is unlikely.
Device migration is also a question. Two patients (0.9%) experienced device “malposition, dislodgement, or movement.” As CyPass position was measured at regular intervals postoperatively with no reported movement other than in these two patients, postoperative migration appears unlikely to be a concern. Further, although movement may occur in the first few weeks after surgery, the angle tissues fibrose readily around the device inlet in the first month, and thus movement after this time seems less likely. This has been our clinical experience as well.
ASCRS created a CyPass Withdrawal Task Force, which recently provided an overview of the results presented by Alcon.1 As per the recommendations of this task force, patients who received the device should be screened with a complete slit-lamp examination including gonioscopy to assess position and look for any contact with the cornea. The task force stated that “numerous conditions and therapeutic interventions can result in ECL, but intervention is generally limited to when clinically apparent or functionally significant changes occur.”1 The task force also concluded that no intervention is likely needed if the device has no rings or one ring visible. If two or three rings are visible, patients are at higher risk of ECL; however, no intervention is likely required if there are no signs of corneal decompensation. More frequent corneal assessment may be required. Of note, even if more than one ring is visible, the device may still be far enough from the peripheral cornea to be safe, as individual angle depth can vary from one patient to the next.
The task force suggested that, if quantification is desired, the physician can obtain a baseline and follow-up ECD and pachymetry, but that there is significant variability in measurements and techniques and clinical examination may be sufficient for monitoring eyes with an indwelling CyPass device. “If corneal decompensation develops and [more than one] ring of the device is visible, the surgeon may consider CyPass repositioning, removal, or proximal end trimming,”1 according to the task force. In our experience, due to firm attachments of the device to surrounding uveal tissue, we do not recommend explantation beyond the first month due to the risk of significant surgical trauma. Instead, the device can be trimmed using microforceps and microscissors when needed.
HOW DO WE ASSESS ECL AFTER SURGERY?
ECL may occur due to surgical trauma at the time of surgery and/or progressively due to chronic endothelial cell trauma or irritation postoperatively. Because these devices and procedures are located near the peripheral cornea, when we measure central ECD (as studies report), some central ECL occurs due to the trauma of the surgery itself. However, it may take time to show further central loss due to a potential peripheral corneal insult. One must also keep in mind when reviewing the available data that a significant confounder in many MIGS studies is concomitant cataract surgery, which in and of itself is a cause of ECL. Very little, if any, data have been published on long-term ECL with cataract surgery alone.
It is important to note that ECD analysis is not a simple measurement, and considerable technical, imaging, and analytical expertise is required to guarantee accuracy. In order to ensure a standardized approach to ECD analysis for FDA trials, the data collected for COMPASS and other MIGS trials underwent examination by a central core laboratory (Cornea Image Analysis Reading Center at Case Western Reserve University). The methodology used for the evaluation and quantification of ECD has been described previously. The process employed permits reproducible and reliable reading of endothelial image quality, ECD, and morphometric analyses.2
RISK/BENEFIT IS WHAT COUNTS IN GLAUCOMA INTERVENTION
When selecting a glaucoma therapy (drops, laser, MIGS, or traditional surgery), with the knowledge that each has its own inherent risks, one must balance the risk-to-benefit ratio. This involves weighing the benefits of the therapy against the risk of permanent, long-term vision loss and blindness from glaucoma, knowing that this is a progressive disease and still one of the most common causes of blindness globally. For the early glaucoma patient, the risk of therapy must be very low, as the risk of serious loss of vision, at least in the short term, is not high. However, for patients with advanced or progressive glaucoma, we are willing to take a hit on risks to counter the higher risk of blindness. We are also more likely to take more procedural risks when the efficacy is greater (and less likely if the efficacy is lower).
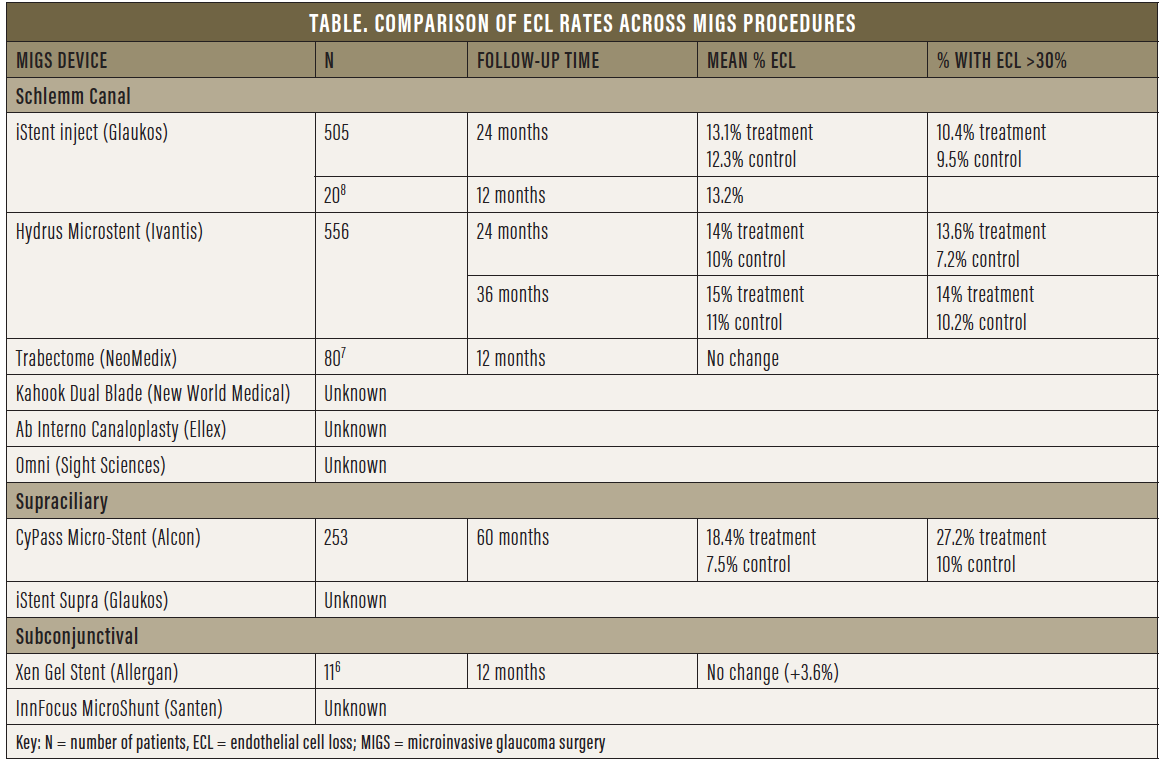
Grouping tube shunts and trabeculectomy with all MIGS procedures would be a fallacy, as would be grouping all MIGS procedures together. MIGS can be categorized based on outflow, whether Schlemm canal- or supraciliary-based versus subconjunctival (Table). Theoretically, canal- or supraciliary-based MIGS procedures, although not as potent, must be safer than traditional surgery, as the target population is mostly low-risk patients with mild to moderate glaucoma who are also undergoing cataract surgery. Typically, the purpose of these surgeries is to increase compliance, decrease drop load, improve the ocular surface, and delay further surgical intervention. Thus, as an early and preventive intervention, the objective is to improve quality of life and delay vision loss, which, from the patient’s perspective, is crucial. Not all MIGS procedures have the same potency or risks. Our safety standards are inherently higher and more stringent for MIGS—particularly canal- or supraciliary-based procedures, which are less potent than subconjunctival procedures.
On the other hand, because traditional surgery is typically performed for advanced or progressive disease, even with known risks such as ECL, we often accept those risks considering the benefit of protection from progressive, permanent glaucomatous loss. Similarly, the threshold for risk tolerance is higher for subconjunctival MIGS procedures, which provide IOP-lowering potency similar to that of traditional filtering surgery, offer potential for improved safety and faster recovery, but carry more risks than canal-or supraciliary-based MIGS procedures.
DO WE HAVE DATA ON ECL AND MIGS?
Each canal-based or supraciliary-based MIGS device varies in how it lies within the angle. The anatomic position of the different devices gives us an idea of their potential effects on ECL. The iStent (Glaukos) is inserted within Schlemm canal with its short nozzle (<300 μm) protruding from the angle far from the endothelium (Figure 4). This is the same ideal position taken by the Hydrus Microstent (Ivantis), with the inlet lying parallel to the iris in continuation with the scaffold placed in the canal (Figure 5).

Figure 4. The iStent (generation 1; top) and iStent inject (generation 2; bottom) views in the angle. iStent dimensions: length = 1 mm, height = 300 μm; iStent inject dimensions: diameter = 230 μm, height = 360 μm
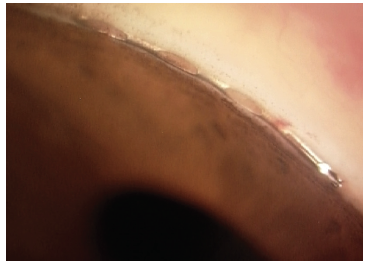
Figure 5. The Hydrus Microstent view in the angle. Dimensions: overall length = 8 mm, major axis = 292 μm, minor axis = 185 μm, outer diameter = 292.1 μm, and inner diameter = 241.3 μm
The CyPass Micro-Stent, due to its stiffness and positioning in the supraciliary space, follows the curvature of the inner sclera and assumes a more vertical orientation in the angle, and thus it is located closer to the peripheral cornea (Figure 6). If the device is placed too anteriorly or migrates, it may come into contact with the cornea. On the other hand, if migration or malpositioning of the iStent or Hydrus occurs, these devices will follow the contour of the canal and remain far from the cornea (of note, migration has not been an issue with these devices). Theoretically, the concerns with CyPass are not the same as those for other canal-based procedures, although long-term data are needed.
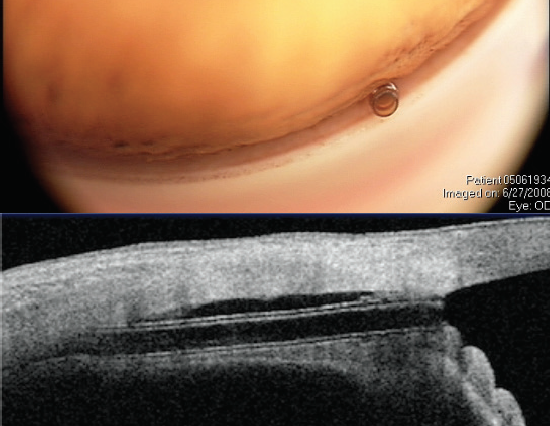
Figure 6. The CyPass Micro-Stent views in the angle (top) and on anterior segment OCT (bottom). Note the ideal deeper position of the implant with the collar below Schwalbe line (and no rings present). Dimensions: overall length = 6.35 mm, outer diameter = 430 μm, and inner diameter = 300 μm
Upon withdrawal of CyPass from the market, the FDA stated: “The FDA has no information indicating similar long-term [ECL] issues with other approved minimally invasive glaucoma surgery devices, but we continue to closely monitor the progress of ongoing postapproval studies for these devices.”3 At the time the FDA approved the iStent, the first MIGS device to receive approval, ECD analysis was not mandated, and thus we have no data on ECL from the iStent pivotal study. More than 400,000 iStents have been implanted to date, with more than 10 years of data, and no known reports of corneal complications due to the device have been reported.
A review of numerous adverse event databases, including the FDA’s Manufacturer and User Facility Device Experience, the European Databank on Medical Devices, and the Australian Database of Adverse Event Notifications, identified no direct associations between corneal decompensation and the iStent. Both the iStent inject (Glaukos) and the Hydrus Microstent have shown no statistically significant differences in ECL at 24 months compared with controls in postoperative adverse event data presented to the FDA. Recent 3-year followup data on the Hydrus Microstent showed ECD counts stable from the 2-year timepoint and no significant difference compared with cataract surgery alone.4,5 ECL assessment for the Hydrus device will continue through 5-year followup.
So far, there are few data with regard to subconjunctival MIGS devices. For the Xen Gel Stent (Allergan; Figure 7), a small nonrandomized, retrospective study of 11 patients showed no change in ECD at 12 months.6 No data are available for the InnFocus MicroShunt (Santen; Figure 8).
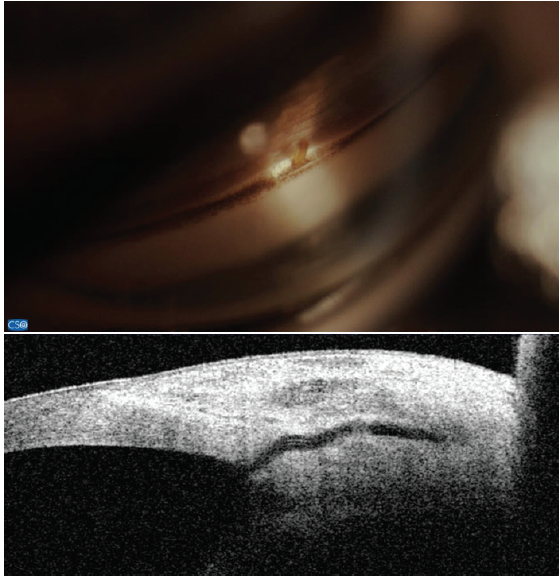
Figure 7. The Xen Gel Stent views in the angle (top) and on anterior segment OCT (bottom). Note the short length in the anterior chamber and the distance from the cornea. Dimensions: overall length = 6 mm, outer diameter = 150 μm (expands to 220 μm when hydrated), and inner diameter = 45 μm
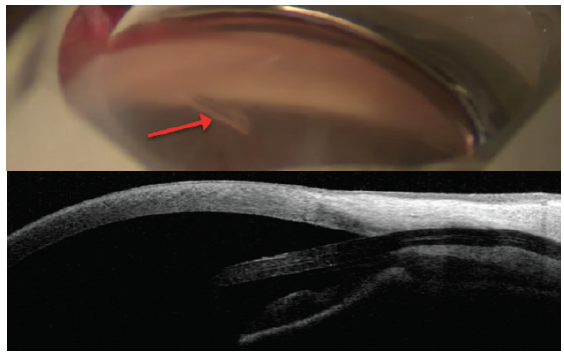
Figure 8. The InnFocus MicroShunt views in the angle (top) and on anterior segment OCT (bottom). Note the distance from the cornea. Dimensions: overall length = 8.5 mm, outer diameter = 350 μm, and inner lumen diameter = 70 μm
Goniotomy- or trabeculotomy-creating procedures also lack short- and long-term ECD data. A single retrospective review (n = 80) of the Trabectome (NeoMedix) showed no statistically significant difference in pre- and postoperative ECL after 12 months. No data on controls were provided.7 As for the Kahook Dual Blade (New World Medical), the Omni (Sight Sciences), and the Ab Interno Canaloplasty procedure (Ellex), no data are currently available, but we would expect minimal ECL aside from the endothelial cell damage that takes place at the time of surgery, given that no device remains in the angle.
IS ECL A CONCERN FOR ALL MIGS PROCEDURES?
With the data currently available, we feel confident that canal-based MIGS procedures do not carry the same risks as the CyPass due to their anatomic positioning. The discussion of subconjunctival MIGS—for which the risk of ECL is unknown—is also different, as the risk-to-benefit ratio for progressive or uncontrolled glaucoma is not the same. Although we must be critical, at this time it does not seem prudent to group all MIGS procedures together and/or to suggest that all have a risk for ECL similar to that of the CyPass Micro-Stent. Further long-term data on all MIGS devices and procedures will be useful going forward to confirm their safety.
The MIGS train has already departed. Challenges are bound to arise as we chase our goals to improve quality of life, address adherence, achieve target IOPs, and prevent blindness in the long run. The bottom line is that we learn from these obstacles to help improve patient outcomes. They are a necessary means to the evolution of glaucoma treatment. Identifying current and future problems is crucial to increasing the success of these procedures. We are in an unprecedented era with a need for high-quality, evidence-based, long-term data (which is a good thing), but this desire for certainty also makes it more difficult to study and bring innovations to the market. We must be mindful of this and balance the risks and benefits and access to therapy for the blinding disease of glaucoma.
The ultimate goal is to customize treatment based on the risk-benefit profile for every patient. The path may be arduous, but the future remains bright.
1. Preliminary ASCRS CyPass withdrawal consensus statement. ASCRS. 2018. http://ascrs.org/CyPass_Statement. Accessed September 12, 2018.
2. Krachmer JH, Mannis MJ, Holland EJ. Cornea: Fundamentals, Diagnosis and Management. 3rd ed. New York: Elsevier; 2011: 177-197.
3. Potential eye damage from Alcon CyPass Micro-Stent used to treat open-angle glaucoma: FDA safety communication. FDA. September 14, 2018. www.fda.gov/MedicalDevices/Safety/AlertsandNotices/ucm620646.htm. Accessed September 14, 2018.
4. Samuelson TW, Chang DF, Marquis R, et al; HORIZON Investigators. A Schlemm canal microstent for intraocular pressure reduction in primary open-angle glaucoma and cataract: the HORIZON study [published online ahead of print June 23, 2018]. Ophthalmology. doi:10.1016/j.ophtha.2018.05.012.
5. Hydrus Microstent. FDA. www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/DeviceApprovalsandClearances/Recently-ApprovedDevices/ucm620440.htm. Accessed September 12, 2018.
6. Fea AM, Spinetta R, Cannizzo PML, et al. Evaluation of bleb morphology and reduction in IOP and glaucoma medication following implantation of a novel gel stent. J Ophthalmol. 2017;2017:9364910.
7. Maeda M, Watanabe M, Ichikawa K. Evaluation of trabectome in open-angle glaucoma. J Glaucoma. 2013;22(3):205-208.
8. Arriola-Villalobos P, Martinez-de-la-Casa JM, et al. Mid-term evaluation of the new Glaukos iStent with phacoemulsification in coexistent open-angle glaucoma or ocular hypertension and cataract. Br J Ophthalmol. 2013;97(10):1250-1255.

